
Uncategorized
Cellular Senescence: What is it?
What is Cellular Senescence?
Cellular senescence is a state in which cells permanently stop dividing while remaining metabolically active, typically triggered by DNA damage or other cellular stressors. First described by Leonard Hayflick in studies of human fetal fibroblasts, senescence distinguishes non-transformed cells from malignant cells, which can replicate indefinitely. Unlike quiescent cells, which can reenter the cell cycle, or terminally differentiated cells, senescent cells are permanently arrested but exhibit unique features, including chromatin reorganization, altered gene expression, and the senescence-associated secretory phenotype (SASP), a pro-inflammatory profile.
The role of senescence is context-dependent, with both protective and harmful effects. It is thought to have evolved as a mechanism to prevent the malignant transformation of damaged cells. However, the accumulation of senescent cells over time contributes to age-related diseases, including cancer, tissue degeneration, and chronic inflammation. Importantly, senescence is not synonymous with aging; while aging reflects a progressive functional decline, senescence occurs throughout life and plays essential roles in embryogenesis, tissue repair, and wound healing. Despite its involvement in aging and pathology, senescence also remains a vital part of normal biological processes.
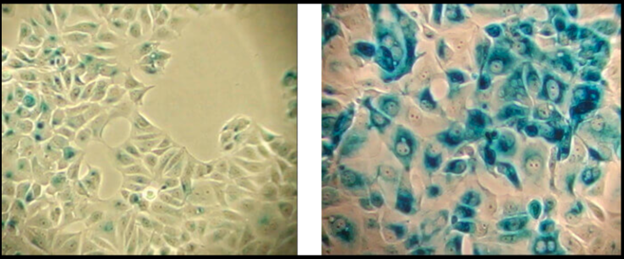
β-Galactosidase staining detects expression of pH-dependent β-galactosidase activity (cells stained in blue), a known characteristic of senescent cells. Normal cells (left panel); Senescent cells (right panel).
Why Does Senescence Occur?
Cellular senescence acts as a critical safeguard in preventing the replication of cells with damaged DNA, thereby playing a pivotal role in protecting the body from tumor development. By halting the cell cycle, senescence ensures that potentially cancerous cells with genomic instability are unable to proliferate, serving as a key anti-tumorigenic mechanism. This protective response is triggered by a variety of cellular stressors, each linked to different types of senescence-inducing stimuli.
- Replicative Senescence occurs due to telomere shortening, which is a natural consequence of repeated cell division. Telomeres, the protective caps at the ends of chromosomes, progressively shorten with each replication. When they become critically short, they signal the cell to enter a state of senescence, preventing further division that could lead to genomic instability.
- DNA Damage-Induced Senescence is initiated when cells experience irreparable damage to their DNA, caused by factors such as radiation, oxidative stress, or exposure to toxic agents. This form of senescence prevents the propagation of mutations that could lead to oncogenesis.
- Oncogene-Induced Senescence arises in response to aberrant oncogenic signaling, such as the activation of mutated proto-oncogenes like RAS or MYC. While these signals drive uncontrolled cell growth in cancer, their early activation in non-transformed cells can paradoxically trigger senescence, acting as a built-in mechanism to suppress tumor formation.
Though senescence plays an essential role in tumor suppression, it is a double-edged sword. In some contexts, the persistence of senescent cells and their inflammatory secretions (senescence-associated secretory phenotype or SASP) can contribute to age-related diseases and chronic inflammation. Nonetheless, its primary role in halting the replication of damaged or dysregulated cells highlights its critical importance in maintaining genomic integrity and reducing the risk of cancer development. This balance between protection and potential pathology underscores the complexity of senescence in biological systems.
Understanding replicative senescence and Hayflick limit
Replicative senescence describes the process by which normal, nonmalignant cells cease dividing after a finite number of cell divisions, a limit often referred to as the Hayflick limit, named after Leonard Hayflick, who first observed the phenomenon in cultured human cells. This cessation of division is driven by telomere shortening, a natural mechanism that prevents genomic instability and uncontrolled proliferation.
Telomeres are repetitive nucleotide sequences at the ends of chromosomes that protect them from degradation and prevent chromosomes from fusing with one another. During each round of DNA replication, a small portion of the telomere sequence is lost due to the inherent limitations of the DNA replication machinery. Over time, telomeres become progressively shorter until they reach a critical length that can no longer safeguard the chromosome.
At this critical threshold, uncapped telomeres are recognized as damaged DNA by the cell’s surveillance mechanisms. This initiates a robust DNA damage response (DDR), which activates pathways such as p53 and p21 to halt the cell cycle, effectively triggering senescence. This state ensures that cells with critically short telomeres do not continue to divide, thereby preventing genomic instability and potential malignant transformation.
While replicative senescence serves as a vital tumor suppressor mechanism, it is also a contributor to aging and age-related diseases. The accumulation of senescent cells over time can lead to tissue dysfunction, driven in part by the senescence-associated secretory phenotype (SASP)—a pro-inflammatory state that disrupts the surrounding cellular environment.
Understanding replicative senescence provides critical insights into aging biology and cancer prevention. Therapies aimed at protecting telomeres, clearing senescent cells, or mitigating SASP effects hold promise for addressing age-related decline and diseases while preserving the anti-cancer benefits of this essential cellular checkpoint.
DNA Damage-Induced Senescence
DNA damage activates a sophisticated cellular response system to maintain genomic integrity and prevent the propagation of damaged cells. The outcome of this response—whether DNA repair, apoptosis (programmed cell death), or senescence (a state of irreversible cell cycle arrest)—depends on the extent of the damage and the physiological context in which it occurs.
When DNA damage is detected, the cell activates the DNA damage response (DDR), a highly conserved signaling cascade that halts cell cycle progression to allow for repair. If the damage is minor, the repair machinery restores the DNA, enabling normal cellular function to resume. However, if the damage is extensive or irreparable, the DDR triggers more drastic outcomes: apoptosis to eliminate the compromised cell, or senescence to permanently arrest its division and prevent potential oncogenic transformation
Senescent cells are characterized by a persistent DDR, sustained in part by chronic activation of ATM (Ataxia Telangiectasia Mutated) and ATR (Ataxia Telangiectasia and Rad3-Related) kinases. These kinases initiate signaling cascades that converge on two critical pathways:
1. p53/p21 Pathway: ATM and ATR activate p53, a tumor suppressor protein, which induces the expression of p21. This cyclin-dependent kinase inhibitor halts the cell cycle, effectively enforcing senescence.
2. p16/pRb Pathway: Independently, p16 also inhibits cyclin-dependent kinases, maintaining the hypo-phosphorylated state of the retinoblastoma protein (pRb), which reinforces the block on cell cycle progression.
Persistent DNA damage and the subsequent induction of senescence can be triggered by various internal and external stressors, including:
- Ionizing Radiation: High-energy radiation damages DNA directly by inducing double-strand breaks.
- Chemotherapeutics: Many cancer therapies work by causing DNA damage, forcing cancer cells into senescence or apoptosis.
- Genotoxic Stress: Exposure to agents that chemically modify DNA can impair replication and transcription, leading to damage accumulation.
- Oxidative Stress: Reactive oxygen species (ROS) generated by metabolic processes or environmental factors can damage DNA, proteins, and lipids, amplifying the DDR.
While senescence serves as a protective mechanism to prevent the replication of damaged cells and potential tumorigenesis, its long-term persistence can have deleterious effects. Senescent cells accumulate with age and secrete pro-inflammatory factors collectively known as the senescence-associated secretory phenotype (SASP), which can contribute to tissue dysfunction, chronic inflammation, and age-related diseases.
Understanding the interplay between DNA damage, repair pathways, and senescence not only highlights its role in cancer prevention but also underscores the potential for therapeutic interventions. Strategies such as targeting senescent cells (senolytics), modulating DDR signaling, or reducing oxidative stress offer promising avenues for mitigating the negative consequences of persistent senescence while preserving its protective functions.
Oncogene-Induced Senescence
Cellular senescence serves as a powerful, cell-autonomous anti-cancer mechanism, particularly in response to oncogenic signaling. This process, known as oncogene-induced senescence (OIS), is a protective cellular response that halts the proliferation of cells experiencing abnormal oncogenic activity, thereby preventing their progression into malignant tumors. By entering senescence, cells with potentially dangerous oncogenic mutations avoid uncontrolled growth, reducing the risk of cancer development.
OIS is triggered by the hyperactivation of oncogenes or the inactivation of tumor suppressor genes, which disrupt normal cellular regulatory mechanisms.
For example:
- H-Ras Oncogene Activation: The expression of H-RAS^V12, a mutated and hyperactive form of the GTPase H-RAS, induces OIS by promoting chronic activation of the p38 MAPK (mitogen-activated protein kinase) pathway. This pathway sustains a persistent DNA damage response (DDR), ultimately enforcing senescence to prevent the cell from dividing uncontrollably.
- Tumor Suppressor Inactivation: Loss of function in genes such as PTEN, a key tumor suppressor that regulates cellular growth signals, can also lead to unregulated oncogenic activity. In response, senescence is induced to arrest growth and maintain cellular integrity.
A significant hallmark of OIS is the replication stress caused by excessive mitogenic signaling. Hyperactive oncogenes accelerate the cell cycle, leading to increased demand on DNA replication machinery. This results in stalled replication forks, which are vulnerable to collapse and generate DNA damage. The subsequent activation of DDR pathways, including ATM and ATR signaling, halts the cell cycle and reinforces the senescence program through p53/p21 and p16/pRb pathways.
While OIS plays a crucial role in tumor suppression, the persistence of senescent cells in the tissue microenvironment can have mixed consequences. On one hand, senescence prevents malignant transformation, but on the other, senescent cells secrete inflammatory factors collectively known as the senescence-associated secretory phenotype (SASP). These pro-inflammatory molecules can alter the surrounding tissue environment, sometimes promoting tumor progression or tissue dysfunction.
Understanding the mechanisms of OIS provides valuable insights into cancer biology and therapeutic opportunities. Strategies to leverage OIS include:
- Enhancing the senescence response to eliminate early-stage tumorigenic cells.
- Using senolytic therapies to clear senescent cells and mitigate SASP-related inflammation.
- Targeting replication stress and DDR pathways selectively in cancer cells to exploit their vulnerabilities.
By deepening our understanding of OIS, researchers aim to refine cancer prevention strategies and develop therapies that maximize its protective effects while minimizing its long-term drawbacks. This duality of OIS emphasizes its critical role as both a safeguard against malignancy and a potential contributor to age-related tissue changes.
Biomarkers of Senescence: How do we measure it?
Cellular senescence is a critical biological process characterized by stable cell cycle arrest, accompanied by profound changes in cell morphology, metabolism, chromatin structure, and gene expression. Senescent cells also adopt the senescence-associated secretory phenotype (SASP), which plays complex roles in tissue health, aging, and disease. However, not all senescent cells display all known biomarkers, and some senescence markers are shared with other cellular states, such as apoptosis or quiescence. As a result, identifying senescent cells requires a comprehensive analysis of multiple biomarkers. Below is an in-depth examination of these defining features.
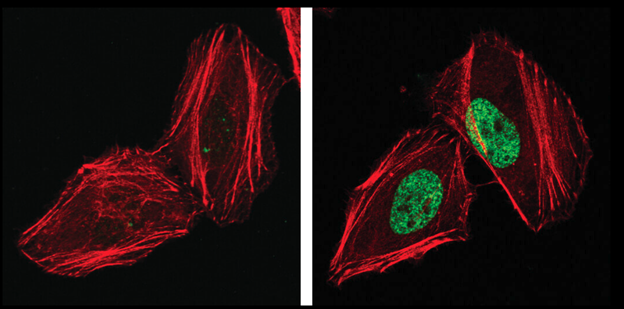
Phospho-Histone H2A.X (Ser139), also known as γ-H2A.X is a commonly used marker of cell senescence (green-stained nucleus in the image on the right).
Stable Cell Cycle Arrest
The hallmark of cellular senescence is a permanent state of cell cycle arrest, distinguishing senescent cells from quiescent cells, which can reenter the cell cycle when stimulated. Senescent cells do not divide in response to any known physiological stimuli, ensuring that damaged or potentially cancerous cells are prevented from proliferating.
This stable arrest is enforced by the activation of key tumor suppressor pathways, namely:
- p53/p21CIP1 Pathway: Activated by DNA damage, this pathway halts cell cycle progression by inhibiting cyclin-dependent kinases.
- p16INK4A/pRb Pathway: p16INK4A inhibits cyclin-dependent kinases, keeping the retinoblastoma protein (pRb) in a hypo-phosphorylated state, which further enforces cell cycle arrest.
While p16INK4A is frequently used as a biomarker for senescence, it is not exclusive to senescent cells. For example, p16INK4A expression can also occur in certain tumor types and cell lines lacking functional pRb, highlighting the need to evaluate additional markers for accurate identification.
Morphological and Metabolic Changes
Senescent cells undergo dramatic physical and functional changes:
- Morphology: Senescent cells often exhibit an enlarged, flattened shape with increased vacuolization. In some cases, they may become multinucleated. These features are indicative of the extensive structural and functional changes associated with senescence.
- Nuclear Envelope Integrity: A hallmark of senescent cells is the loss of lamin B1, a key component of the nuclear envelope. This loss disrupts nuclear integrity and contributes to chromatin reorganization.
- Mitochondrial Dysfunction: Senescent cells accumulate dysfunctional mitochondria, leading to elevated production of reactive oxygen species (ROS). These ROS exacerbate oxidative stress and contribute to the senescence phenotype.
- Lysosomal Activity: Increased lysosomal content and activity are prominent in senescent cells, often measured through elevated β-galactosidase activity at pH 6.0, a widely used biomarker for senescence.
These metabolic alterations not only reflect the senescent state but also contribute to the pro-inflammatory phenotype associated with SASP, further amplifying their impact on tissue health.
Loss of Lamin B1 (shown here in green) is a marker of cellular senescence.
Chromatin Reorganization and Altered Gene Expression
One of the most distinctive features of senescent cells is chromatin remodeling, which results in widespread changes in gene expression:
- Senescence-Associated Heterochromatin Foci (SAHF): These heterochromatic structures silence genes that promote proliferation, such as E2F target genes like cyclin A. SAHF are visually identifiable through bright DAPI staining and immunoreactivity for markers such as macroH2A, heterochromatin protein 1 (HP1), and di-/tri-methylated histone H3 (H3K9Me2/3).
- Variability of SAHF: While SAHF formation is a hallmark of some senescent cells, it is not universal; some cells undergo senescence without forming these structures.
These chromatin changes are essential for reinforcing the stable cell cycle arrest of senescent cells and contribute to their unique transcriptional profiles.
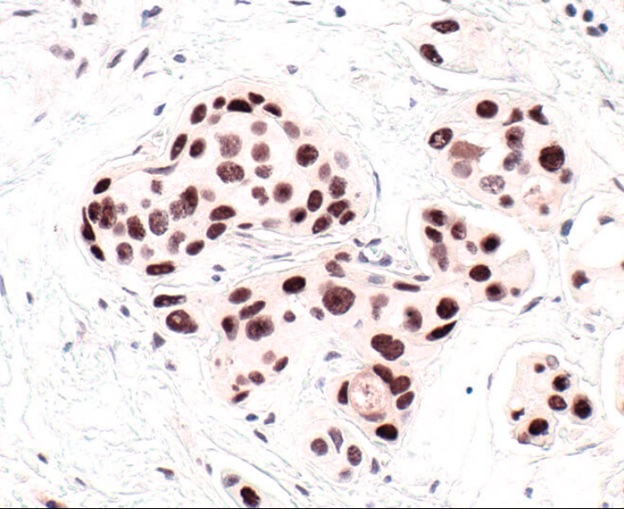
HP1 is a commonly used marker of cellular senescence. Immunohistochemical analysis of HP1 expression in paraffin-embedded human breast carcinoma using HP1 alpha antibody.
DNA Damage and Persistent DNA Damage Response (DDR)
DNA damage is a central trigger of senescence, particularly when the damage is irreparable:
- Persistent DDR: Chronic activation of DNA damage response pathways, such as ATM and ATR kinases, leads to the stabilization of p53 and the formation of DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS).
- Telomere Dysfunction-Induced Foci (TIFs): When telomeres become critically short or uncapped, they trigger a specific DDR, halting cell division to prevent genomic instability.
- γ-H2A.X as a Marker: Phosphorylated H2A.X (γ-H2A.X) is a key marker of double-stranded DNA breaks and persistent DNA damage in senescent cells.
This persistent DDR not only enforces senescence but also contributes to the pro-inflammatory signaling associated with SASP.
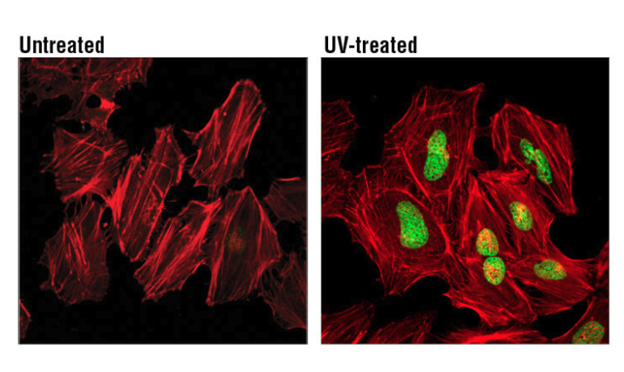
H2A.X is a commonly used marker of cell senescence. Confocal immunofluorescent analysis of HeLa cells, untreated (left), treated with UV (100 mJ/cm2) using Phospho-Histone H2A.X (Ser139) Mouse mAb (green). Actin filaments were labeled with DyLight 554 Phalloidin (red).
Senescence-Associated Secretory Phenotype (SASP)
The SASP is a defining feature of senescent cells, characterized by the secretion of cytokines, chemokines, growth factors, and proteases:
- Protective Roles: SASP components recruit immune cells to eliminate senescent cells, thereby reducing the risk of malignant transformation and supporting tissue repair.
- Pathogenic Roles: In chronic senescence, SASP can promote tumor progression through angiogenesis, extracellular matrix remodeling, or epithelial-mesenchymal transition (EMT). It can also drive systemic inflammation, contributing to age-related diseases such as cancer, arthritis, and neurodegeneration.
The composition of the SASP varies based on the cell type and the senescence-inducing stimulus, reflecting its complex and context-dependent role in tissue biology.
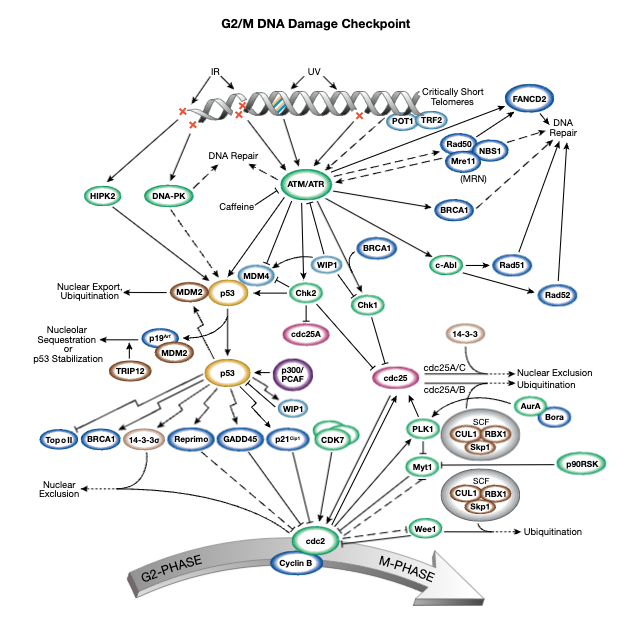
Signaling Pathways for Senescence
Triggers of senescence ultimately converge on the p53/p21CIP1 and p16INK4A/pRb tumor suppressor pathways which induce cell cycle arrest.
Cell Cycle Arrest
Stable cell cycle arrest is a defining feature of senescence, and occurs in response to telomere shortening, oncogenic signaling, and DNA damage. Cell cycle arrest is mediated by the activation of either the p53/p21CIP1 or p16INK4A/phospho-Rb tumor suppressor pathways. Interestingly, mutations in pathway components are commonly observed in different types of cancer.
Persistent DNA damage response (DDR) due to chronic genomic stress or telomere attrition leads to activation of the tumor suppressor p53, which in turn activates p21CIP1 to initiate cell cycle arrest. Upstream of p53 are the kinases ATR and ATM, which act through Chk1 and Chk2, respectively to activate p53 as part of the DDR. Once activated by p53, p21CIP1 inhibits cyclin dependent kinases (CDKs) which prevents cell cycle progression through the G1 to S phase checkpoint. In addition, CDK inhibition blocks phosphorylation of Retinoblastoma tumor suppressor protein (Rb) which prevents E2F-mediated transcription of proliferation-promoting genes. Inhibition of phosphorylated Rb is also directly mediated by the activation of p16INK4A which prevents CDK4 and CDK6 mediated phosphorylation of Rb and downstream E2F-mediated transcription.
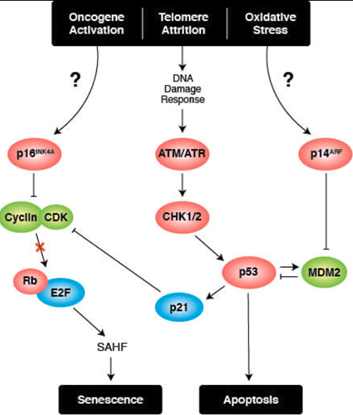
Transcriptional Regulation of Cellular Senescence
Sources:
Product available for research use only:

